
Ensaios clínicos são estudos de pesquisa que visam determinar se uma estratégia médica, tratamento ou dispositivo é seguro para uso ou consumo por seres humanos.
Esses estudos também podem avaliar a eficácia de uma abordagem médica para condições específicas ou grupos de pessoas.
Em geral, eles adicionam ao conhecimento médico e fornecem dados confiáveis para auxiliar na tomada de decisões e diretrizes de cuidados com a saúde.
Para garantir a segurança dos participantes, os testes começam com pequenos grupos e examinam se um novo método causa algum dano ou efeitos colaterais insatisfatórios. Isso ocorre porque uma técnica que é bem sucedida em um laboratório ou em animais pode não ser segura ou eficaz para seres humanos.
Fatos rápidos sobre ensaios clínicos
- Os ensaios clínicos visam descobrir se uma estratégia médica, tratamento ou dispositivo é seguro e eficaz para o uso ou consumo de humanos.
- Os ensaios consistem em quatro fases e podem enfocar: tratamento, prevenção, diagnóstico, triagem, cuidados de suporte, pesquisa em serviços de saúde e ciência básica.
- Uma equipe de pesquisa provavelmente incluirá médicos, enfermeiros, assistentes sociais, profissionais de saúde, cientistas, gerentes de dados e coordenadores de ensaios clínicos.
- A participação pode envolver riscos e benefícios. Os participantes devem ler e assinar o documento de “consentimento informado” antes de ingressar em um teste.
- Os riscos são controlados e monitorados, mas a natureza dos estudos de pesquisa médica significa que alguns riscos são inevitáveis.
O que são ensaios clínicos?
O principal objetivo dos ensaios clínicos é a pesquisa. Ensaios são projetados para adicionar ao conhecimento médico relacionado ao tratamento, diagnóstico e prevenção de doenças ou condições.

Os estudos seguem rígidos padrões científicos e diretrizes que visam:
- proteger os participantes
- fornecer resultados confiáveis e precisos
Os ensaios clínicos em humanos ocorrem nos estágios finais de um processo de pesquisa longo, sistemático e completo.
O processo geralmente começa em um laboratório, onde novos conceitos são desenvolvidos e testados.
Testes em animais permitem aos cientistas ver como a abordagem afeta um corpo vivo.
Finalmente, o teste humano é realizado em grupos pequenos e depois maiores.
Os ensaios podem ser realizados para:
- Avaliar uma ou mais intervenções de tratamento para uma doença, síndrome ou condição, como medicamentos, dispositivos médicos ou abordagens para cirurgia ou terapias
- Avaliar maneiras de prevenir uma doença ou condição, por exemplo, através de medicamentos, vacinas e mudanças no estilo de vida
- Avaliar uma ou mais intervenções de diagnóstico que possam identificar ou diagnosticar uma determinada doença ou condição
- Examine os métodos de identificação para reconhecer uma condição ou fatores de risco para essa condição
- Explorar procedimentos de cuidados de apoio para melhorar o conforto e a qualidade de vida das pessoas com uma doença crónica
O resultado de um ensaio clínico pode identificar se uma nova estratégia, tratamento ou dispositivo médico:
- tem um efeito positivo no prognóstico do paciente
- causa danos imprevistos
- não tem benefícios positivos ou tem efeitos negativos
Os ensaios clínicos podem fornecer informações valiosas sobre a relação custo-eficácia de um tratamento, o valor clínico de um teste de diagnóstico e como um tratamento melhora a qualidade de vida.
Tipos de ensaio clínico
Todos os ensaios clínicos têm um objetivo principal. Estes podem ser divididos nas seguintes categorias:
- Tratamento: Testando novos tratamentos, novas combinações de medicamentos ou novas abordagens para cirurgia ou terapia
- Prevenção: Examinando formas de melhorar a prevenção ou recorrência de doenças através, por exemplo, de medicamentos, vitaminas, vacinas, minerais e mudanças no estilo de vida
- Diagnóstico: Encontrar técnicas e procedimentos de teste aprimorados para diagnosticar doenças e condições
- Triagem: Testando o melhor método de identificação de certas doenças ou condições de saúde
- Cuidados de suporte: Investigando procedimentos para melhorar o conforto e a qualidade de vida de pacientes com uma condição crônica
- Pesquisa de serviços de saúde: Avaliação da entrega, processo, gestão, organização ou financiamento de cuidados de saúde
- Ciência básica: examinando como funciona uma intervenção
Por que os ensaios clínicos são importantes?
Ensaios clínicos ajudam a melhorar e avançar os cuidados médicos. Os estudos fornecem evidências factuais que podem ser usadas para melhorar o atendimento ao paciente.
A pesquisa clínica só é realizada quando os médicos desconhecem elementos como:
- se uma nova abordagem funciona de forma eficaz em humanos e é segura
- que tratamentos ou estratégias funcionam com mais sucesso para certas doenças e grupos de indivíduos
Como os ensaios clínicos funcionam?
Vários elementos estão envolvidos na configuração, execução e acompanhamento de um estudo clínico.
Protocolo de ensaios clínicos

Um julgamento segue um plano abrangente ou protocolo. Um protocolo é a descrição escrita de um ensaio clínico.
Inclui os objetivos, o design e os métodos do estudo, o histórico científico relevante e a informação estatística.
A informação chave a incluir pode ser:
- o número de participantes
- que é elegível para participar
- que testes serão dados e com que frequência
- tipos de dados a serem coletados
- a duração do estudo
- informações detalhadas sobre o plano de tratamento
Evitando o viés
Os pesquisadores devem tomar medidas para evitar preconceitos.
Viés refere-se a escolhas humanas ou outros fatores que não estão relacionados ao protocolo, mas que podem afetar os resultados do estudo.
Etapas que podem ajudar a evitar distorções são grupos de comparação, randomização e mascaramento.
Grupos de comparação
A maioria dos ensaios clínicos usa grupos de comparação para comparar estratégias e tratamentos médicos. Os resultados mostrarão se um grupo tem um resultado melhor que o outro.
Isso geralmente é realizado de duas maneiras:
- Um grupo recebe um tratamento existente para uma condição e o segundo grupo recebe um novo tratamento. Os pesquisadores então comparam qual grupo tem melhores resultados.
- Um grupo recebe um novo tratamento e o segundo grupo recebe um placebo, um produto inativo que se parece com o produto de teste.
Randomization
Ensaios clínicos com grupos de comparação frequentemente usam randomização. Os participantes são alocados para grupos de comparação por acaso, e não por escolha. Isso significa que quaisquer diferenças observadas durante um teste serão devidas à estratégia usada e não devido a diferenças pré-existentes entre os participantes.
Mascarando ou cegando
Mascarar ou cegar ajuda a evitar preconceitos ao não informar os participantes ou os pesquisadores sobre o tratamento que os participantes receberão.
Único cego: É quando os participantes ou pesquisadores não sabem, de qual grupo é qual.
Double blind: Isto é quando ambos os participantes e pesquisadores não estão cientes.
Fatores Confusionais
Um confundidor pode distorcer a verdadeira relação entre duas ou mais características.
Por exemplo, pode-se concluir que as pessoas que carregam um isqueiro são mais propensas a desenvolver câncer de pulmão porque carregar um isqueiro causa câncer de pulmão. Fumar é um fator de confusão neste exemplo.
As pessoas que carregam um isqueiro são mais propensas a serem fumantes, e os fumantes são mais propensos a desenvolver câncer de pulmão, mas algumas pessoas podem carregar um isqueiro para outros fins.
Não levar isso em consideração pode levar a conclusões falsas.
Quem está na equipe de pesquisa?
Um investigador principal, que geralmente é um médico, conduzirá cada estudo clínico.
A equipe de pesquisa pode incluir:
- doutores
- enfermeiros
- trabalhadores sociais
- profissionais de saúde
- cientistas
- gerenciadores de dados
- coordenadores de ensaios clínicos
Onde os ensaios clínicos são conduzidos?
A localização dependerá do tipo de estudo e de quem está organizando.
Alguns locais comuns incluem:
- hospitais
- universidades
- centros médicos
- consultórios médicos
- clínicas comunitárias
- sites de pesquisa financiados pelo governo federal e financiados pela indústria
Quanto tempo duram os testes?
Isso depende do que está sendo estudado, entre outros fatores. Algumas tentativas duram dias, enquanto outras continuam por anos.
Antes de se inscrever em um julgamento, os participantes serão informados quanto tempo deve durar.
Projetado e organização
Existem diferentes tipos de estudo e diferentes formas de organizá-los. Aqui estão alguns tipos de estudo.
Estudos observacionais
Estudos de coorte e estudos de caso-controle são exemplos de estudos observacionais.
Estudo de coorte

Um estudo de coorte é um estudo observacional no qual a população do estudo, ou coorte, é selecionada.
As informações são coletadas para determinar quais assuntos têm:
- uma característica particular, como um grupo sanguíneo que se acredita estar relacionado ao desenvolvimento da doença em questão
- exposição a um fator que pode estar ligado a uma doença, por exemplo, tabagismo
Um indivíduo pode ser escolhido porque fuma. Eles podem então ser seguidos a tempo para ver a probabilidade de desenvolver uma doença, em comparação com outras pessoas.
Esse tipo de estudo é usado para estudar o efeito de fatores de risco suspeitos que não podem ser controlados experimentalmente, como o impacto do tabagismo no câncer de pulmão.
As principais vantagens dos estudos de coorte são:
- A exposição é medida antes do início da doença e, portanto, é provável que seja imparcial em termos de desenvolvimento da doença.
- Exposições raras podem ser investigadas por seleção adequada de coortes de estudo.
- Múltiplos desfechos – ou doenças – podem ser estudados para qualquer exposição.
- A incidência da doença pode ser calculada nos grupos expostos e não expostos.
As principais desvantagens dos estudos de coorte são:
- Eles tendem a ser caros e demorados, especialmente se forem conduzidos de maneira prospectiva, o que significa avançar.
- Mudanças no estado de exposição e nos critérios diagnósticos ao longo do tempo podem afetar a classificação dos indivíduos de acordo com a exposição e o status da doença.
- Pode haver viés de informação no resultado final porque o status de exposição do sujeito é conhecido.
- Perdas no seguimento podem apresentar viés de seleção.
Estudos de controle de caso
Um estudo de caso-controle pode distinguir fatores de risco para uma condição médica particular.
Pesquisadores comparam pessoas com uma condição e aquelas sem ela. Retrocedendo no tempo, eles identificam como os dois grupos diferem.
Os estudos de caso-controle são sempre retrospectivos – olhando para trás – porque começam com o resultado e, em seguida, voltam para investigar as exposições.
As principais vantagens dos estudos de caso-controle são:
- Os resultados podem ser obtidos rapidamente.
- O estudo pode ser realizado com um mínimo de financiamento ou patrocínio.
- Eles são eficientes para investigar doenças raras ou doenças com um longo período de indução.
- Uma ampla gama de possíveis fatores de risco pode ser examinada.
- Exposições múltiplas podem ser estudadas.
- Eles exigem poucos sujeitos do estudo.
As principais desvantagens dos estudos controlados por casos são:
- Dados de incidência não podem ser gerados.
- Eles estão sujeitos a viéses.
- Pode ser difícil obter medidas precisas e imparciais de exposições passadas se a manutenção de registros for inadequada ou não confiável. Isso é chamado de viés de informação.
- A seleção de controles pode ser problemática. Isso pode introduzir um viés de seleção.
- A sequência cronológica entre a exposição e a doença pode ser difícil de identificar.
- Eles não são apropriados para examinar exposições raras, a menos que a exposição seja responsável por uma grande porcentagem de casos.
Estudo de caso-controle aninhado
Em um estudo caso-controle aninhado, os grupos – casos e controles – vêm da mesma população de estudo, ou coorte.
À medida que a coorte é seguida, os casos que surgem tornam-se os “casos” no estudo de caso-controle. Os participantes não afetados da coorte se tornam os “controles”.
Estudos de caso-controle aninhados são menos onerosos e consomem menos tempo quando comparados a um estudo de coorte.
A incidência e as taxas de prevalência da doença podem ocasionalmente ser projetadas a partir de um estudo de coorte de caso-controle aninhado. Isso não é possível em um estudo de caso-controle simples, pois o número total de indivíduos expostos e os tempos de acompanhamento são geralmente desconhecidos.
As principais vantagens dos estudos de caso-controle aninhados são:
- Eficiência: nem todos os participantes da coorte exigem testes de diagnóstico.
- Flexibilidade: Permitem testar hipóteses que não foram previstas quando a coorte foi planejada.
- Redução do viés de seleção: Casos e controles são amostrados da mesma população.
- Redução do viés de informação: A exposição ao fator de risco pode ser avaliada com o pesquisador cego ao status do caso.
A principal desvantagem é que os resultados têm menor autoridade, devido ao pequeno tamanho da amostra.
Estudo ecológico
Um estudo ecológico analisa a relação entre exposição e resultado da população ou comunidade.
Categorias comuns de estudo ecológico incluem:
- comparações geográficas
- análise de tendência temporal
- estudos de migração
As principais vantagens dos estudos ecológicos são:
- Eles são baratos, pois dados de saúde coletados rotineiramente podem ser utilizados.
- Eles são menos demorados do que outros estudos.
- Eles são simples e fáceis de entender.
- O efeito de exposições que são medidas em grupos ou áreas – como dieta, poluição do ar e temperatura – pode ser investigado.
As principais desvantagens dos estudos ecológicos são:
- Erros de dedução conhecidos como falácia ecológica podem ocorrer. Acontece quando os pesquisadores tiram conclusões sobre os indivíduos com base apenas na análise de dados de grupo.
- A exposição a relações de resultados é difícil de detectar.
- Há falta de informação sobre fatores de confusão.
- Pode haver diferenças sistemáticas entre as áreas em como as exposições são medidas.
Estudos experimentais
Além de estudos observacionais, existem também estudos experimentais, incluindo estudos de tratamento.
Ensaios controlados randomizados

Um ensaio clínico randomizado (ECR) aleatoriamente aloca indivíduos para receber ou não uma determinada intervenção.
Um dos dois tratamentos diferentes será usado, ou um tratamento e um placebo.
Este é o tipo de estudo mais eficaz para identificar qual tratamento funciona melhor. Reduz a influência de variáveis externas.
As principais vantagens dos ECRs são:
- Não há preconceito consciente ou subconsciente por parte do pesquisador. Isso essencialmente garante validade externa.
- Variáveis de confusão, como idade, sexo, peso, nível de atividade e assim por diante, podem ser canceladas, desde que o grupo de amostras seja grande o suficiente.
As principais desvantagens dos ECRs são:
- Eles são demorados.
- Eles podem ser caros.
- Eles exigem grandes grupos de amostras.
- Eventos raros podem ser difíceis de estudar.
- Ambos os erros estatísticos falsos positivos e falsos negativos são possíveis.
Ensaio clínico adaptativo
Um método de design adaptativo é baseado nos dados coletados. É flexível e eficiente. Modificações podem ser feitas no teste e nos procedimentos estatísticos dos ensaios clínicos em andamento.
Quasi-experimento
Estudos quase experimentais, ou “não randomizados”, incluem uma ampla gama de estudos de intervenção que não são randomizados. Este tipo de ensaio é frequentemente utilizado quando um ECR não é logisticamente viável ou ético.
Hierarquia de evidência
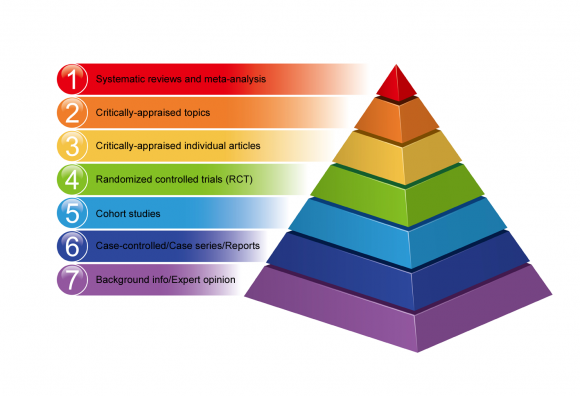
As hierarquias de evidência possibilitam classificar vários métodos de pesquisa de acordo com a validade de suas descobertas.
Nem todos os projetos de pesquisa são iguais em termos de risco de erro e viés em seus resultados. Alguns métodos de pesquisa fornecem melhores evidências do que outros.
Abaixo está um exemplo da hierarquia da medicina baseada em evidências na forma de uma pirâmide, variando de uma qualidade inferior de evidências na parte inferior até evidências de alta qualidade no topo.
Fases de um ensaio clínico
Estudos de pesquisa médica são divididos em diferentes fases, chamadas fases. Para o teste de drogas, estes são definidos pelo FDA.
Os estudos de fase inicial investigam a segurança de um medicamento e os efeitos colaterais que podem causar. Testes posteriores testam se um novo tratamento é melhor que um tratamento existente.
Ensaios da fase 0: farmacodinâmica e farmacocinética
A fase 0 é uma fase exploratória que ajuda a fornecer informações clínicas para um novo medicamento em uma fase anterior.
Esta fase:
- é realizado no início da fase 1
- envolve exposição humana muito limitada
- não tem intenção terapêutica ou diagnóstica, limitando-se aos estudos de triagem e microdose
Ensaios da Fase 1: Rastreio de segurança
Após a fase 0, existem mais quatro fases de testes em seres humanos. Estes geralmente se sobrepõem. As fases 1 a 3 ocorrem antes de uma licença ser concedida.
As diretrizes da fase 1 envolvem:
- entre 20 e 80 voluntários saudáveis
- verificação dos efeitos colaterais mais freqüentes da droga
- descobrir como a droga é metabolizada e excretada
Ensaios da Fase 2: Estabelecendo a eficácia
Se os estudos da fase 1 não revelarem níveis de toxicidade inaceitáveis, os estudos da fase 2 podem começar.
Isso involve:
- entre 36 e 300 participantes
- coleta de dados preliminares sobre se a droga funciona em pessoas com uma determinada doença ou condição
- ensaios controlados para comparar aqueles que recebem a droga com pessoas em situação semelhante que estão recebendo uma droga diferente ou um placebo
- avaliação de segurança contínua
- estudos de efeitos colaterais de curto prazo
Ensaios da fase 3: confirmação final da segurança e eficácia
Se a fase 2 confirmou a eficácia de um medicamento, a FDA e os patrocinadores discutirão como conduzir estudos de larga escala na fase 3.
Isso envolverá:
- entre 300 e 3.000 participantes
- recolha de mais informações sobre segurança e eficácia
- estudos de diferentes populações
- examinar várias dosagens para determinar o melhor valor de prescrição
- usando a droga em combinação com outras drogas para determinar a eficácia
Após essa fase, as informações completas sobre o novo medicamento são submetidas às autoridades de saúde.
Reunião de revisão
Se o FDA aprovar o produto para comercialização, são realizados estudos de exigência e comprometimento pós-marketing.
A FDA usa esses estudos para coletar informações adicionais sobre segurança, eficácia ou uso otimizado do produto.
Novo pedido de drogas

Um patrocinador de medicamentos preencherá uma Solicitação de Novo Medicamento (NDA) para pedir à FDA que considere a aprovação de um novo medicamento para comercialização nos EUA.
Um NDA inclui:
- todos os dados animais e humanos
- análise de dados
- informações sobre o comportamento de drogas no corpo
- detalhes de fabricação
A FDA tem 60 dias para decidir se deseja arquivá-lo para ser revisado.
Se eles decidirem arquivar o NDA, a equipe de revisão do FDA é designada para avaliar a pesquisa do patrocinador sobre a segurança e a eficácia do medicamento.
Os seguintes passos devem então acontecer.
Rotulagem de medicamentos: O FDA revisa a rotulagem profissional do medicamento e confirma que informações apropriadas são compartilhadas com consumidores e profissionais de saúde.
Inspeção de instalação: O FDA inspeciona as instalações onde o medicamento será fabricado.
Aprovação de medicamentos: os revisores da FDA aprovam a solicitação ou emitem uma carta de resposta.
Ensaios da Fase 4: Estudos durante as vendas
Os ensaios da fase 4 ocorrem após o medicamento ter sido aprovado para comercialização. Eles são projetados para incluir:
- mais de 1.000 pacientes
- experiência abrangente na avaliação da segurança e eficácia do novo medicamento em um grupo maior e subpopulações de pacientes
- comparação e combinação com outros tratamentos disponíveis
- avaliação dos efeitos colaterais a longo prazo da droga
- detecção de eventos adversos menos comuns
- custo-efetividade da terapia medicamentosa em comparação com outras terapias tradicionais e novas
Relatório de segurança
Depois que o FDA aprova uma droga, o estágio de pós-comercialização começa. O patrocinador, geralmente o fabricante, envia atualizações periódicas de segurança para o FDA.
Quem patrocina ensaios clínicos?
Ensaios clínicos e pesquisas podem custar centenas de milhões de dólares. Grupos que financiam ensaios podem incluir:
- empresas farmacêuticas, de biotecnologia e de dispositivos médicos
- centros médicos acadêmicos
- grupos e fundações voluntárias
- Instituto Nacional de Saúde
- departamentos do governo
- médicos e provedores de saúde
- indivíduos
Quem pode participar?
O protocolo define quem é elegível para participar de uma avaliação.
Os possíveis critérios de inclusão podem ser:
- ter uma doença ou condição específica
- ser “saudável”, sem condições de saúde
Os critérios de exclusão são os fatores que excluem algumas pessoas de participar de um estudo.
Os exemplos incluem idade, sexo, um tipo específico ou estágio de uma doença, histórico de tratamento anterior e outras condições médicas.
Possíveis benefícios e riscos
Participar de ensaios clínicos pode trazer benefícios e riscos para os participantes.
Os possíveis benefícios dos ensaios clínicos incluem o seguinte:
- Os participantes têm acesso a novos tratamentos.
- Se um tratamento for bem sucedido, os participantes estarão entre os primeiros a se beneficiar.
- Os participantes que não estão no grupo recebendo um novo tratamento podem receber o tratamento padrão para a condição específica, que pode ser tão boa ou melhor que a nova abordagem.
- A saúde é acompanhada de perto e apoiada por uma equipe de provedores de saúde.
- As informações coletadas de ensaios clínicos acrescentam conhecimento científico, podem ajudar outras pessoas e, em última análise, melhorar os cuidados com a saúde.
Possíveis riscos incluem:
- O cuidado padrão para uma condição particular pode às vezes ser melhor do que a nova estratégia ou tratamentos sendo estudados.
- A nova abordagem ou tratamento pode funcionar bem para alguns participantes, mas não necessariamente para outros.
- Pode haver efeitos colaterais inesperados ou imprevisíveis, especialmente em estudos de fase 1 e fase 2 e com abordagens como terapia gênica ou novos tratamentos biológicos.
- O seguro de saúde e os provedores de saúde nem sempre cobrem o atendimento ao paciente e os custos para aqueles que participam de ensaios clínicos.
O que significa dar consentimento?

O documento de consentimento informado explica os riscos e os benefícios potenciais de participar de um estudo clínico.
Elementos que devem aparecer no documento incluem, entre outros:
- objetivo da pesquisa
- riscos previsíveis de desconforto
- possíveis benefícios
Os participantes devem ler atentamente o documento de consentimento, decidir se querem se inscrever e assinar antes de poderem ser incluídos no estudo.
Os ensaios clínicos são seguros?
A FDA trabalha para garantir que qualquer pessoa que esteja pensando em participar de um teste tenha acesso a todas as informações confiáveis de que precisa para fazer uma escolha informada, incluindo informações sobre os riscos.
Embora os riscos para os participantes sejam controlados e monitorados, alguns riscos podem ser inevitáveis, devido à natureza dos estudos de pesquisa médica.
Como os participantes são protegidos?

A segurança dos participantes é uma questão de alta prioridade. Em todos os ensaios, a supervisão científica e os direitos dos pacientes contribuem para sua proteção.
A boa prática clínica (GCP) visa assegurar que os procedimentos éticos e apropriados sejam seguidos nos ensaios.
A conformidade com o GCP fornece ao público a confiança de que a segurança e os direitos dos participantes estão protegidos.
Tem como objetivo:
- para proteger os direitos, a segurança e o bem-estar dos participantes
- garantir que os dados coletados sejam confiáveis, tenham integridade e sejam de qualidade
- fornecer diretrizes e padrões para a condução da pesquisa clínica
As fundações do GCP foram expostas pela primeira vez em 1947. Os principais pontos foram que, durante qualquer ensaio, os pesquisadores devem garantir:
- participação voluntária
- consentimento informado
- minimização de risco
Com o tempo, os acréscimos variaram desde o estabelecimento de proteção adicional para populações vulneráveis até o fornecimento de orientação a organismos que realizam pesquisas.
Direitos do paciente
As formas de proteger os direitos do paciente incluem o seguinte:
O consentimento informado é o processo de fornecer aos participantes de estudos clínicos todos os fatos sobre o estudo. Isso acontece antes de os participantes concordarem em participar e durante o curso do teste. O consentimento informado inclui detalhes sobre os tratamentos e testes que podem ser recebidos e os possíveis benefícios e riscos.
Outros direitos: O documento de consentimento informado não é um contrato; os participantes podem se retirar do estudo a qualquer momento, independentemente de o teste estar completo ou não.
Direitos e proteção para crianças: Um pai ou responsável legal deve dar o consentimento legal se a criança tiver 18 anos ou menos. Se um teste envolver um risco maior que o mínimo, ambos os pais devem dar permissão. Crianças com idade acima de 7 anos devem concordar em se envolver em ensaios clínicos.
Como faço para encontrar um ensaio clínico?
Informações sobre ensaios clínicos atuais podem ser encontradas aqui.





